With a $1 million grant from the Hereditary Disease Foundation (HDF), a team of top researchers led by Ricardo Mouro Pinto, Ph.D., of Harvard Medical School, is deploying CRISPR to target genetic modifiers of Huntington’s disease.
In brain cells, these modifiers accelerate or slow so-called somatic expansions in HD.
The Mouro Pinto Lab at Harvard-affiliated Massachusetts General Hospital (MGH) specializes in research on these expansions. Scientists describe this process as the tendency of the mutant, expanded, disease-causing huntingtin gene to keep expanding abnormally. This causes the dysfunction – and possibly also death – of brain cells, leading to HD symptoms (click here to read more). As scientists now see somatic expansion as a key driver of HD, research on this process has burgeoned.
Investigators have identified the modifier genes affecting expansion through deep research in human data. In speeding or slowing somatic expansion, those genes can hasten or delay disease onset. Many academic and biopharma groups, including the Mouro Pinto Lab, are investigating modifiers as potential drug targets.
The Mouro Pinto Lab is focusing on how one modifier gene, MLH3, acts harmfully as a “scissors” in HD. (By convention, human MLH3 gene is always capitalized and italicized; in contrast, the non-human gene is rendered as Mlh3.)
In an August 7 interview at his lab, Dr. Mouro Pinto described an experimental drug his team is testing in mice to deactivate the MLH3 scissors, a potential first step in seeking a treatment in people.
“I'm extremely optimistic about our approach,” Dr. Mouro Pinto said. “We can see what our drug does to somatic expansions. Can it slow it down? And our preliminary data is very encouraging in the sense that we can do that.”
On August 10, Dr. Mouro Pinto provided an update on the project at HDF’s HD2024: Milton Wexler Biennial Symposium. Announced in October 2023, the foundation’s two-year Transformative Research Award supports his team’s research of “therapeutic targeting of somatic CAG expansions with CRISPR base editing.” A group of anonymous donors funds the award.
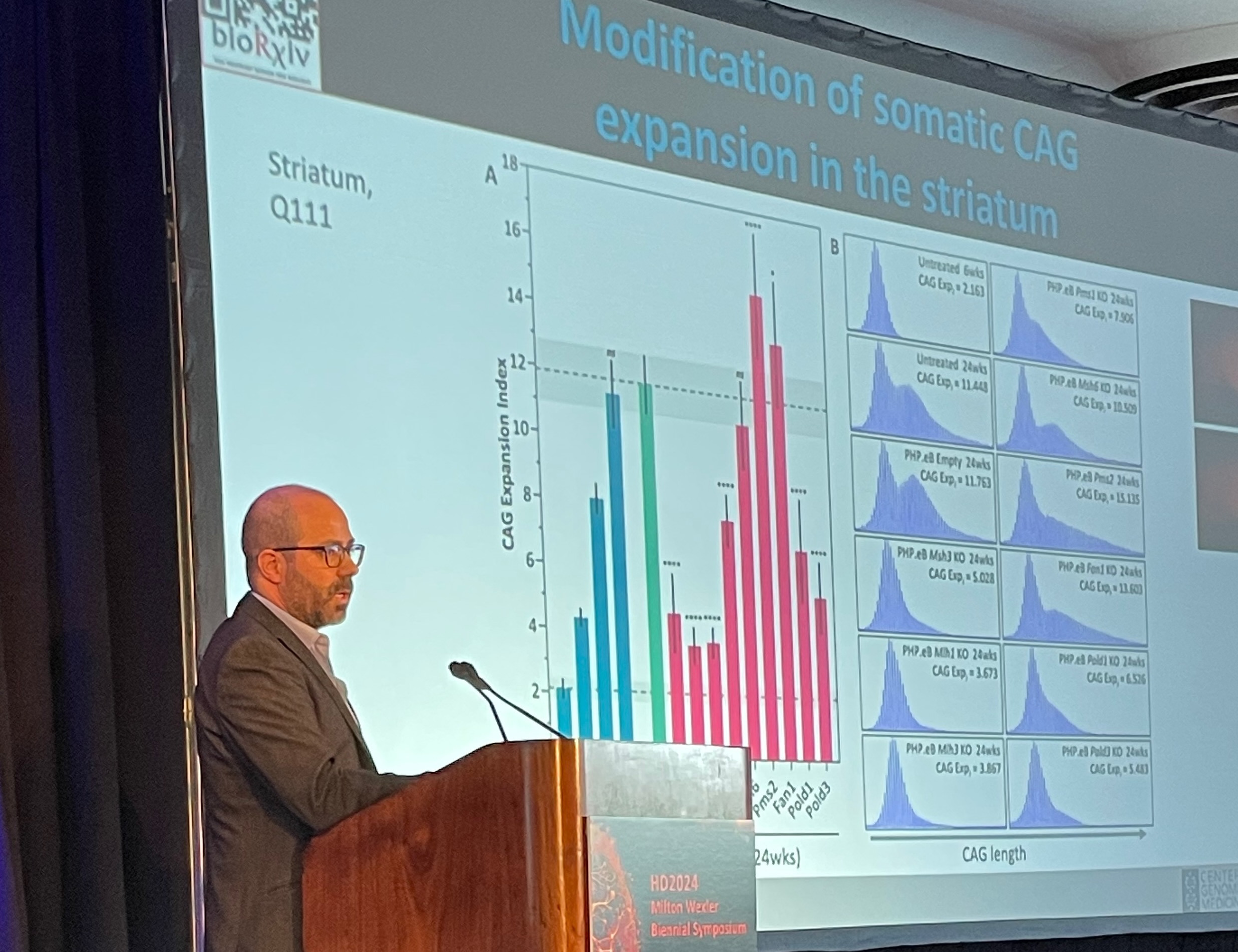
Above, at the HDF symposium, Dr.
Mouro Pinto displays a slide demonstrating how his lab’s experimental drug
slowed somatic expansion in the striatum of mouse brains. In humans, the
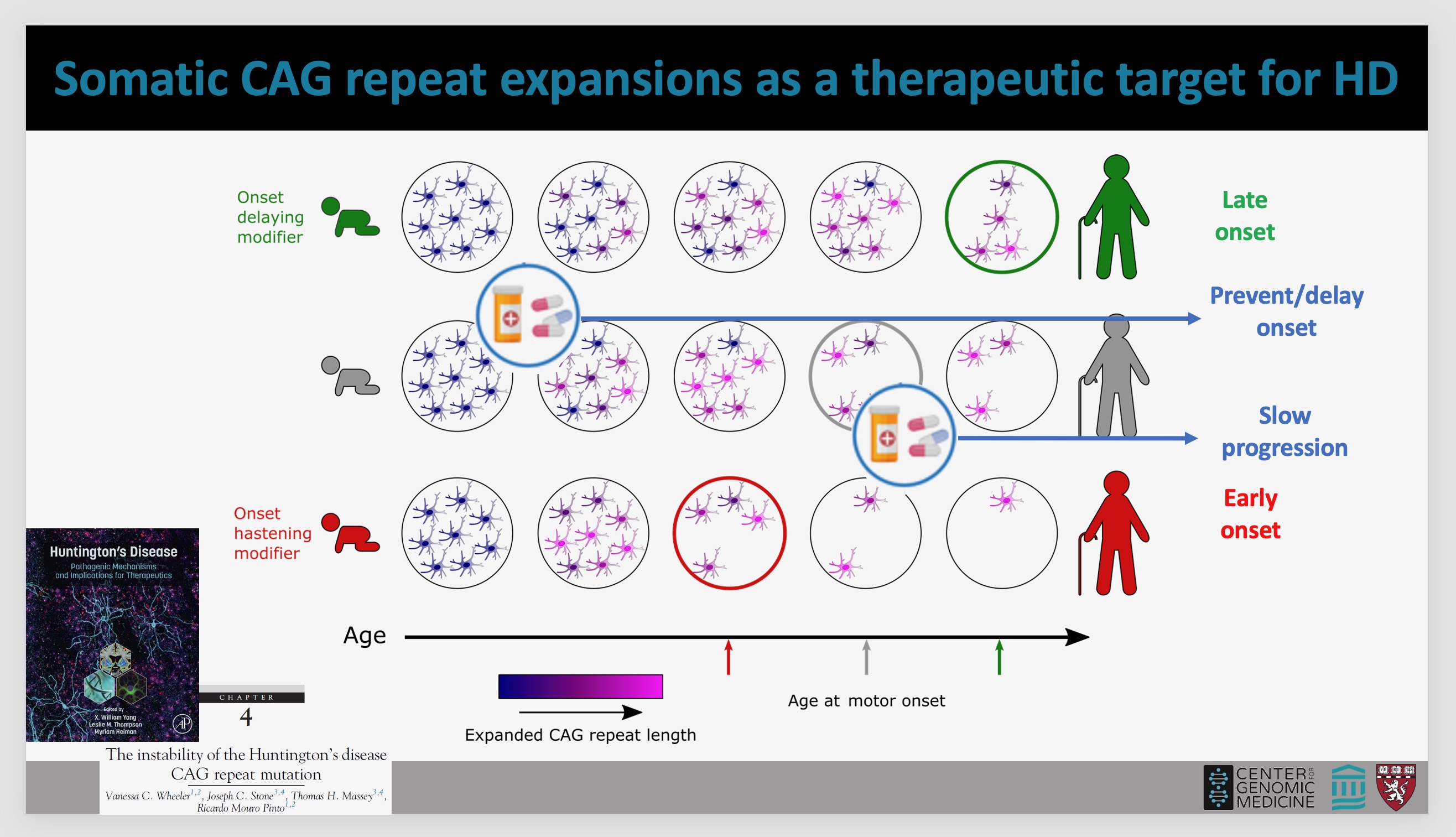
striatum is one of the areas most affected by HD. Below, a Mouro Pinto slide
illustrates how a drug could impact somatic expansion and therefore prevent or
delay HD onset, or slow progression of the disease in humans (photo of Dr. Mouro
Pinto by Gene Veritas, aka Kenneth P. Serbin, image of slide courtesy of Dr.
Mouro Pinto). (To make it larger, click on an image.)
Innovative collaborators
Seven innovative labs are collaborating with the Mouro Pinto team.
James Gusella, Ph.D., whose Harvard lab discovered the huntingtin gene in 1993, is one of three co-investigators on the project.
Gusella’s team has developed the human cell line that “allows us to model somatic instability so we can test the drugs,” Dr. Mouro Pinto explained. That cell line has the expansions of the segment of the DNA code CAG (cytosine, adenine, and guanine), identified in 1993 as the underlying cause of HD.
At the symposium the HDF awarded Gusella the Leslie Gehry Prize for Innovation in Science, including $100,000 for research and a plaque with a small sculpture by the renowned architect Frank Gehry.
Specialists in somatic expansion
A native of Porto, Portugal, Dr. Mouro Pinto received his Ph.D. in molecular genetics at Brunel University in England in 2010, focusing on Friedreich’s ataxia, a debilitating genetic neuromuscular disorder. Along with HD, Friedreich’s is one of more than 50 repeat expansion disorders. In Friedreich’s and others, somatic expansion also plays a role. Scientists also refer to this process as somatic instability.
From 2010-2015, Dr. Mouro Pinto worked as a postdoctoral fellow in the lab of Vanessa Wheeler, Ph.D., an associate professor of neurology at Harvard, MGH researcher, and pioneer in the study of somatic instability. The Wheeler lab is collaborating with the Mouro Pinto group on the HDF project.
For Portuguese-speaking advocates, in 2022 I interviewed Dr. Mouro Pinto in his native tongue, discussing his work and outlook for HD therapies. We spoke at the 17th HD Therapeutics Conference, sponsored by CHDI Foundation, Inc., the largest private funder of HD research and a backer of the Mouro Pinto Lab.
On August 6, the HDF and MGH co-hosted a tour of the Mouro Pinto Lab for about 20 HDF officials, donors, and HD family members. Dr. Wheeler participated, too. I took part at the invitation of the HDF. At the start, Dr. Mouro Pinto presented an overview of the HDF project.

Above, before the tour of his lab, Dr. Mouro Pinto presents a slide demonstrating the path of his scientific studies. Below, Dr. Mouro Pinto explains the purpose of a PCR (polymerase chain reaction) workstation, where they isolate and make billions of copies of the CAG repeat so they can study it in more detail (photos by Gene Veritas).

A key task: measuring the expansion
In 2016 Dr. Mouro Pinto won the first Berman-Topper Family HD Career Development Fellowship from the Huntington’s Disease Society of America. That funding allowed him to conduct research demonstrating that modifier genes can speed or slow somatic expansion in mice, he said.
According to Dr. Mouro Pinto, his lab’s more recent examination of postmortem tissue from HD patients revealed that somatic expansion had occurred in about 30 different brain regions. In all, 50 different tissues were examined, showing different rates of somatic expansion in the liver, muscle, kidney, and others. Some of the fastest expansion was in the brain, he added.
To measure a drug’s effect in a clinical trial, brain biopsies are currently not an option, Dr. Mouro Pinto pointed out. However, measuring somatic expansion in blood, liver, and cerebrospinal fluid, which bathes the brain, could serve as biomarkers. A biomarker is a sign of a disease or effect of a drug. He noted that other researchers are already investigating somatic expansion in the blood.
“What is the right tissue, bio fluid to obtain from the patient?” Dr. Mouro Pinto asked. “And then what is the right test to be sensitive and accurate? So those two things are actually other aspects of research in our lab that we're trying to develop.”
Having those biomarkers will be crucial if the potential CRISPR drug reaches a human clinical trial, he observed.
CRISPR: a one-time, permanent edit of the gene
CRISPR stands for “clustered regularly interspaced short palindromic repeats,” a strand of RNA that, when activated by an enzyme, can edit DNA. Bacteria evolved this technique to defend against viruses.
Jennifer Doudna, Ph.D., and Emmanuelle Charpentier, Ph.D., won the 2020 Nobel Prize in Chemistry for their work in identifying and understanding CRISPR. Click here to read more about CRISPR, its potential for treating HD, and its powerful implications for the future of humanity.
Scientists have now used CRISPR to edit human genes in labs and in clinical trials that have resulted in drugs approved by the U.S. Food and Drug Administration (FDA). In December 2023 the FDA approved two CRISPR drugs for sickle cell disease, an inherited disorder that primarily affects people of African descent.
This first wave of CRISPR clinical trials has started with the “lowest-hanging fruit,” diseases that “affect the liver or the eye or the ear,” Dr. Mouro Pinto pointed out. The brain is far more difficult to research, so it has been challenging to address brain diseases with CRISPR drugs, he said.
“You're doing it at the level of the DNA and that causes a permanent change,” he emphasized. “Once you treat and you introduce the change you made, that will stay in that cell forever.”
A CRISPR clinical trial “will look, essentially, the same as any other HD trial,” he explained. “You'll need to collect samples. And you'll need to conduct a battery of physical tests, cognitive tests and behavioral tests. It will still be evaluated exactly by the same standards as any other clinical trial.”
In contrast with most other HD drug approaches, CRISPR has a key, beneficial difference. “It’s a one-time treatment,” Dr. Mouro Pinto said.
An ‘amazing toolbox’
Dr. Mouro Pinto underscored that in addition to serving as a drug, CRISPR has provided an “amazing toolbox” for less costly and more efficient and precise lab research.
In contrast with the old, very expensive process of breeding large numbers of mice over years, CRISPR “has accelerated the rate of research tremendously” and dramatically reduced the numbers needed, he said. With CRISPR, they can much more quickly pursue tasks such as making mutations in mice or screening a large number of genes to see which might modify somatic expansion, he said. The lab’s paper on this topic will be published in Nature Genetics.
The lab also can deliver a CRISPR reagent to a young mouse, transforming it into a model for study and producing results in a few weeks.
A unique ‘humanized Mlh3 mouse’
Use of CRISPR enabled a “really critical” step in the HDF project: creation of what Dr. Mouro Pinto described as a “humanized Mlh3 mouse,” a unique research step. To prepare the potential CRISPR drug for testing in mice for efficacy against HD and safety, the lab introduced into the animals a small sequence of human DNA from the MLH3 gene.
The scientists have crossed these mice with engineered HD mice. After a few generations downstream, this will result in mice with the expanded and the humanized modifier gene, which will be tested with the CRISPR reagent to see if it stops somatic expansion, Dr. Mouro Pinto continued.
The crossing was also necessary to assure that the humanized segment of DNA itself does indeed experience somatic instability, because the goal of the research is to stop instability, he added.
Another key step will involve measuring the CRISPR reagent’s impact on somatic expansion, irregular movements, and behavioral symptoms in the mice, Dr. Mouro Pinto said. For this stage, the lab will have the key assistance of the “extremely experienced” Cathleen Lutz, Ph.D., M.B.A., vice president of The Jackson Laboratory Rare Disease Translational Center, he noted.
‘Promoting the flavor without the scissors’
As it develops greater precision, the Mouro Pinto Lab has found that one of two MLH3 variants has the scissors that cause harmful genetic cutting. That gets closer to solving the HD puzzle.
In a February HDF webinar about the project, Dr. Mouro Pinto explained that in people without HD, the MLH3 variant without scissors – the good variant – is present.
He said that he is confident that promoting expression (activation) of the good variant over the bad would be better tolerated as a treatment than simply turning off the bad. He described this approach as “promoting the flavor without the scissors, as opposed to completely getting rid of the protein” that results from the gene.
“In a mouse that doesn’t have the scissors, you completely stabilize the repeat,” Dr. Mouro Pinto stated in the webinar. “We know that the scissors component of this protein is essential for promoting CAG expansions.”
Furthermore, in human HD cells the team not only reduced but eliminated the bad version of MLH3.
They achieved this using a technique known as base editing.
Dr. Mouro Pinto noted that with standard CRISPR editing, a sequence of DNA can be broken up, potentially causing unwanted effects. In contrast, in base editing, no breakage occurs, because scientists edit the DNA by simply changing a letter of the genetic code, for example, from A (adenine) to G (guanine), or C (cytosine) to T (thymine).
Significantly, his team edited both copies of the MLH3 gene and completely shifted expression from the “bad” version towards only making the “good” “scissor-less” version of MLH3 protein. As a result, the experiment completely stabilized the CAG repeat (i.e. the CAG stopped expanding in edited cells), Dr. Mouro Pinto stressed.
There are two bases because, as Dr. Mouro Pinto reminded, every cell has two copies of each gene – a copy from each parent.
Partnering on more precise gene editing
Only a few projects have started exploring base editing for HD, and most are happening in research labs such as his, Dr. Mouro Pinto said.
To maximize the benefits for HD patients, the HDF project will seek to improve on this editing.
Recognizing the many other labs are examining directly targeting the CAG expansion, Dr. Mouro Pinto believes that, instead, deactivating a modifier gene such as the MLH3 scissors is a safer and easier strategy.
On this aspect, the Mouro Pinto Lab will partner with co-investigator David Liu, Ph.D., of both Harvard and the Massachusetts Institute of Technology (MIT). Dr. Liu invented both base editing and prime editing.
As Dr. Mouro Pinto pointed out in our interview, base editing allows for more “precise base changes.” In the HDF webinar, he noted that this method prevents breaking up of the DNA.
Harvard’s Benjamin Kleinstiver, Ph.D., another innovative co-investigator, has engineered CRISPR enzymes that “literally can go anywhere in the genome,” Dr. Mouro Pinto added. This enables the team to target any sequence of DNA it needs to, he said.
The lab continues to seek improvements in its enzyme to “achieve highest specificity and maximum efficacy,” he said.
The potential path to a clinical trial
“It's too early” to gauge whether the experimental CRISPR reagent can undergo testing in humans and have a “therapeutic impact,” Dr. Mouro Pinto told me, noting that four key questions must be answered in mice first. The lab aims to answer them in the coming months.
He described the outcome needed in the series of mouse experiments: “Did we change the DNA? Yes. Okay. Did we change which version of MLH3 is made? Yes. Okay. Next. Did we reduce the CAG instability? Yes. And then do we have any impact on HD symptoms?”
Currently the lab is working on the first step, with “promising” indications so far, Dr. Mouro Pinto said.
If the project proves successful, other, distinct projects testing the reagent in other animals, such as nonhuman primates, would follow, Dr. Mouro Pinto explained. The HDF grant does not include funds for those steps.
Emphasizing safety, avoiding unwanted edits
Dr. Mouro Pinto underscored that the team strives to find the safest CRISPR drug possible.
In line with more stringent FDA standards and bioethical concerns regarding gene editing, the project must carry out “due diligence” to avoid “very serious adversity.” That includes so-called off-target effects of gene editing, in which a gene such as one for cancer is accidentally activated or turned off, Dr. Mouro Pinto cautioned.
“It is not uncommon for these drugs to have some activity in unwanted regions of the genome,” he explained. “We need to spend a lot of time looking for unwanted modifications.”
The CRISPR agent is “not yet ready for the clinic,” Dr. Mouro Pinto added.
He noted that the project is not doing edits in the sex cells; offspring therefore cannot inherit any genetic changes.
Finding the best way to deliver the cargo
The lab has no name yet for the experimental reagent, that is, its CRISPR enzyme.
“This is still an experimental reagent,” Dr. Mouro Pinto stressed. “I don't want to create false expectations. We are primarily putting effort into making sure that our cargo is good, that it's really doing what we want.”
The “cargo,” the potential drug, needs to be delivered safely and effectively into the brain and to the right cells, Dr. Mouro Pinto said. A common strategy in gene therapy is using a virus, specifically, an adeno-associated virus (AAV).
The Mouro Pinto Lab is using an AAV that works well in mice but not possible for humans, he said. As part of the HDF project, the team is searching for the ideal delivery system, which could be an AAV, a lipid nanoparticle, or extracellular vesicle, he continued. All three are tiny.
“There are many people now working on AAVs that you inject systemically,” Dr. Mouro Pinto said. “You give it into a vein, they go everywhere in your body including crossing the blood-brain barrier and entering the brain.” They can reach “almost every single neuron in the brain,” he added.
The blood-brain barrier is a membrane that protects the brain from harmful substances and germs.
Another project collaborator, Benjamin Deverman, Ph.D., of Vector Engineering and Harvard and MIT, has greatly improved the ability of AAVs to cross the blood-brain barrier in humans. In May, these critical findings for solving brain disorders were published in Science magazine.
“It's going to unlock this sort of roadblock that we have with delivery,” Dr. Mouro Pinto said of this breakthrough.
Lipid nanoparticles also can be injected into the blood. Some researchers are exploring oral administration of extracellular vesicles.
These vehicles pose less burden on clinical trial participants and patients in comparison with other methods, such as spinal taps or direct injection into the brain, a “complex surgical procedure,” Dr. Mouro Pinto observed.
“We're a little bit agnostic to the delivery strategy,” Dr. Mouro Pinto said, noting that the science of these delivery methods is evolving rapidly. By the time the project concludes in October 2025, “there might be a variety of different delivery options that we may want to consider.”
“Synergizing” with other HDF awardees
Dr. Mouro Pinto sees “opportunities to synergize” with the team that also received a 2023 Transformative Research Award, under the leadership of Beverly Davidson, Ph.D., of the University of Pennsylvania and Jang-Ho Cha, M.D., Ph.D., the chief scientific officer of Latus Biosciences. Latus focuses on precision delivery of gene therapy. An expert in AAVs, Dr. Davidson presented the team’s work at the HDF symposium.
Titled “Advancing gene therapies for HD” and focusing on AAVs, that project could potentially provide a delivery system for the CRISPR reagent, Dr. Mouro Pinto said.

Dr. Beverly Davidson presenting her team’s work on AAVs at the 2024 HDF symposium (photo by Gene Veritas).
Getting to market, looking beyond HD
In the event of the CRISPR reagent’s success in the lab, MGH will assist in commercializing it, Dr. Mouro Pinto said.
The hospital could license the technology to a biopharma company or, as in the case of the Davidson-Cha project, to start a company like Latus to bring the drug through a clinical trial and to market.
“We're open to those conversations and we've been fortunate to have a very collaborative interaction with industry partners so far,” Dr. Mouro Pinto told me.
The problem of somatic expansion “is shared across a large number of repeat expansion diseases,” he observed. “Individually, they're rare diseases. Collectively, they're not a rare disease. They actually affect a large number of patients around the world.”
“If our hypothesis is correct, the therapeutic benefit will not be limited to HD patients,” he concluded.
Disclosure: the Hereditary Disease Foundation covered my travel expenses to tour the Mouro Pinto Lab and attend the 2024 symposium.
Sadly, Michael McCabe, a 62-year-old Boston HD man who told his story at this year’s HDF symposium, died suddenly on September 12. Donations in Michael’s memory are suggested to the Huntington’s Disease Society of America.

Gene Veritas (left) and Dr. Mouro Pinto in the MGH lab (personal photo)

